- A
- A
- A
- ABC
- ABC
- ABC
- А
- А
- А
- А
- А
Scientists Create Uniquely Stable Trimeric Model of Coronavirus Spike Transmembrane Domain
ISTOCK
A team of Russian scientists, including HSE MIEM researchers, have presented a 3D model of SARS-CoV-2 S-protein transmembrane (TM) domain. Previously, the TM domain had only been believed to anchor the S-protein in its viral membrane without being involved in rearrangement and fusion with the host cell. Yet according to recent studies, the TM domain appears to have a function in the transmission of genetic information, but its role is not yet fully understood. The researchers believe that the model they have created can contribute to a better understanding of viral mechanisms and potentially lead to the development of novel antiviral drugs. The study has been published in the International Journal of Molecular Sciences.
Scientists describe viruses as a non-cellular form of life and even place them on the borderline between living and non-living organisms. This is because viruses do not have a metabolism of their own and can only replicate within a living host cell. This feature is reflected in the viral structure: the outer shell of a virus contains specific proteins that recognise target cell receptors and facilitate viral attachment. In coronavirus, this function is mainly performed by S-proteins, or spike proteins, that can be seen in microphotography as a characteristic solar corona around the virus.
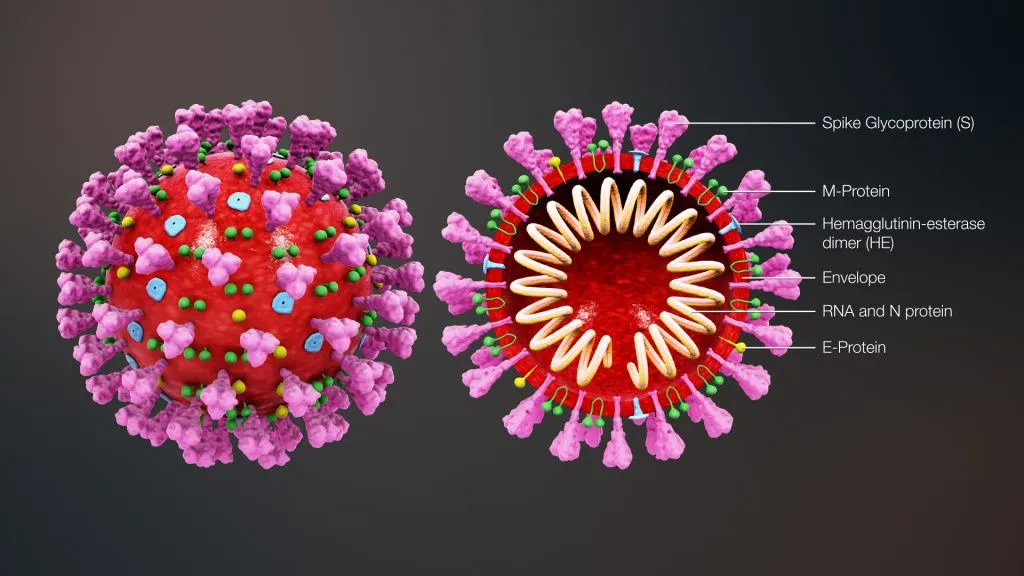{kind=link}
On the target cell surface, the spike (S) protein of the virus interacts with the angiotensin-converting enzyme (ACE), also responsible for regulating blood pressure and maintaining water-electrolyte balance in humans. This is the way for the virus to evade the body's defence mechanisms and attach to host cells.
Understanding viral mechanisms is essential for the development of vaccines and drugs. Therefore, the S-protein’s extra-membrane regions responsible for cell recognition and attachment have been extensively studied. However, the spike protein’s transmembrane domain remains less explored. This fragment of the protein is buried in the viral envelope. A team of scientists from Russia including HSE biophysicists examined this section and created a 3D model of the S-protein's transmembrane domain.
{kind=link}
The spike protein is made up of three identical polypeptide chains that are arranged and attached to each other in a unique manner. When the virus enters the body, the outer portion of the S-protein binds to a receptor on the cell surface. The S-protein is cleaved by proteases, which are proteins found in our body, to reveal previously concealed areas. The structure of the S-protein changes, it becomes embedded in the cellular membrane and, much like cables, pulls the membrane towards its counterpart, the viral envelope. The membranes thus merge to form a channel through which the viral RNA, or genetic material, can penetrate the host cell.
It was during the pandemic that scientists began to notice how the transmembrane domain, which is a small section of the spike protein with a spring-like (helix) shape, is involved in these processes. Previously, it was believed that the TM domain was only necessary for anchoring the S-protein in the viral membrane and did not merit a detailed investigation. The current assumption is that, due to the subtler aspects of its structure, the TM domain may directly contribute to the spike protein's mechanism of action and should be incorporated into relevant models. But creating such a model can be difficult, the study authors note.
When a protein contains membrane-associated regions, recreating its structure using experimental methods can be challenging. Once the transmembrane domain is removed from its natural membrane environment, it loses its spatial structure, making it impossible to understand how exactly the molecule is organised in its functionally active state.
To address this issue, the researchers relied on template-based modelling. This procedure is often employed when a protein’s structure has not yet been determined experimentally. The template acts as a structural framework, onto which the amino acid sequence of the protein is fitted. In order to pick such a template, available structures of TM trimers were examined and their physicochemical properties evaluated both at the level of individual monomers and within a trimer. In addition to this, surfaces of the spike TM domain monomers most likely to be on the helix/helix interface were identified taking into account the results from a program predicting the structure of TM dimers, previously developed by the authors. The template was then chosen, in which individual monomers resembled those in spike TM domain and helix/helix interfaces matched the ones predicted. The model thus obtained was further refined in the course of molecular dynamics simulations in a lipid bilayer, after which the final model was tested likewise, and its stability was confirmed.
To date, this is the only tightly packed, stable model of the S-protein's TM domain. With assistance from our colleagues at the laboratories of the RAS Institute of Bioorganic Chemistry, we will investigate the structural and dynamic behaviour of TM domains experimentally. This is crucial for our understanding of the role of TM- and membrane-proximal domains in the process of membrane fusion and subsequent RNA transmission. By using computer models to investigate this process, one can effectively develop measures to combat viruses. For instance, by altering the characteristics of the interactions of spike protein regions with the membrane, one can create molecules that slow the virus down. This will prevent the virus from spreading as rapidly, making it far less dangerous.
Roman Efremov
Professor, School of Applied Mathematics, MIEM HSE
IQ
Text author: Ekaterina Korchagina
Roman Efremov
Professor, School of Applied Mathematics, MIEM HSE